病毒是地球上数量最多的生物实体,其中细菌病毒(即噬菌体)约有1031个类群,从海洋到陆地再到人体几乎都是它们的栖息地。研究者将病毒视为调节人类生态系统的重要成员,人体内主要包括真核病毒和噬菌体,包括双链DNA (double-stranded DNA, dsDNA),单链DNA (single-stranded DNA, ssDNA)和RNA病毒。随着对病毒研究的广泛开展,“病毒组”与“病毒组学”的概念也应运而生,这些术语分别涵盖了栖息在生态系统中的所有病毒及其基因组和对它们的研究(Lefkowitz EJ, et al, 2017)。根据病毒不同的特征进行分类,包括病毒的宿主范围;病毒的形态学;病毒的基因组大小;病毒的核酸组成成分以及病毒的致病性。虽然所有的性状在病毒分类学的确定中都很重要,但目前利用平均核苷酸同源性(ANI)和系统发育关系进行序列比较被视为定义和区分病毒群类的主要标准。
此外,病毒也被发现潜伏在人类细胞内,如人类内源性逆转录病毒(human endogenous retroviruses, HERVs)等。一部分病毒已经失去了重新激活的能力(例如某些HERVs),另一部分可以重新激活但作为原病毒保留很长一段时间,其它部分则呈现周期性的动态循环(周期性产生病毒粒子并频繁感染) 。另一方面,噬菌体通过与我们体内细菌群落的相互作用,从而可以参与调节人体菌群,对细菌的部分功能基因进行储存,并通过与机体免疫系统的互作,促进免疫系统的成熟。已有大量研究致力于描述病毒群落的特征及它在塑造微生物群方面的作用,然而,不同于细菌和真菌,噬菌体或真核病毒并没有通用的标记基因可以将其作为一个整体进行研究,因此,并不能通过标签序列扩增子测序的手段进行相应的病毒群落解析。
1991年Schmidt(Schmidt TM, et al, 1991)首次提出的环境基因组学(Environmental genomics)被视为宏基因组学的前身,当时Schmidt等构建了世界上第一个海洋生物样本DNA的噬菌体文库,并从中发现了15种全新的细菌核酸序列。1998年,Handelsman等(Handelsman J, et al, 1998)提出研究特定环境样本中遗传物质的总和的课题,并将特定小生境中全部微小生物遗传物质的总和定义为宏基因组(Metagenome) 。2004年Handelsman (Handelsman J, et al, 2004)完整阐述了宏基因组学(Metagenomics)的概念,即以某一特定环境样本中的微生物群体基因组为研究对象,以功能基因筛选和序列测定分析为研究手段,以微生物多样性、种群结构、进化关系、功能活性、相互作用及其与环境之间的关系为研究目的的一种新的微生物研究方法。
宏病毒组学(Viral Metagenomics)是宏基因组学的一个分支,但与传统的宏基因组学概念不同,它是在宏基因组学概念的基础上,结合病毒自身的特点,将宏基因组学方法应用到病毒学领域而形成的。2002年,Breitbart等(Breitbart M, et al, 2002)将宏基因组学方法应用于海洋病毒群落的研究,发现噬菌体为海水中主要病毒组,这一研究标志着宏病毒组学正式应用于科学研究。
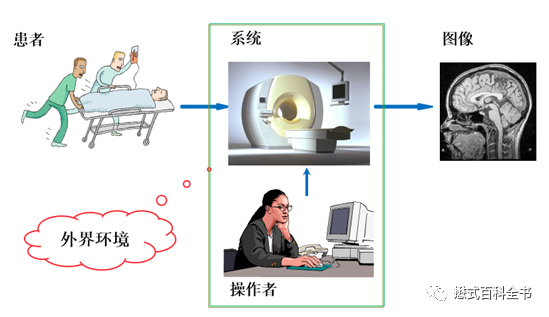
简而言之,宏病毒组学就是应用特殊方法把特定环境中所有病毒与其他微生物分开,然后提取的病毒核酸,用高通量技术进行病毒核酸测序,依托现有数据库进行相应的比对工作,并运用软件分析处理后最终得到研究样品中病毒群落的组成信息。
生物信息分析
介绍宏病毒组分析的常用软件之前,首先我们来看一下宏病毒组的基本分析流程。这里需要说明的是基于病毒颗粒分离技术及NGS测序平台的宏病毒组学研究还处于起步阶段,分析功能模块新的思路和技术层出不穷,但另一个层面,宏病毒组是宏基因组的一个分支,基本大的分析思路方面与宏基因组还是有着不小的相似之处。归纳一下,宏病毒组主要包括一下几个方面的分析内容:
下面是易基因科技的宏病毒组的基本分析流程:
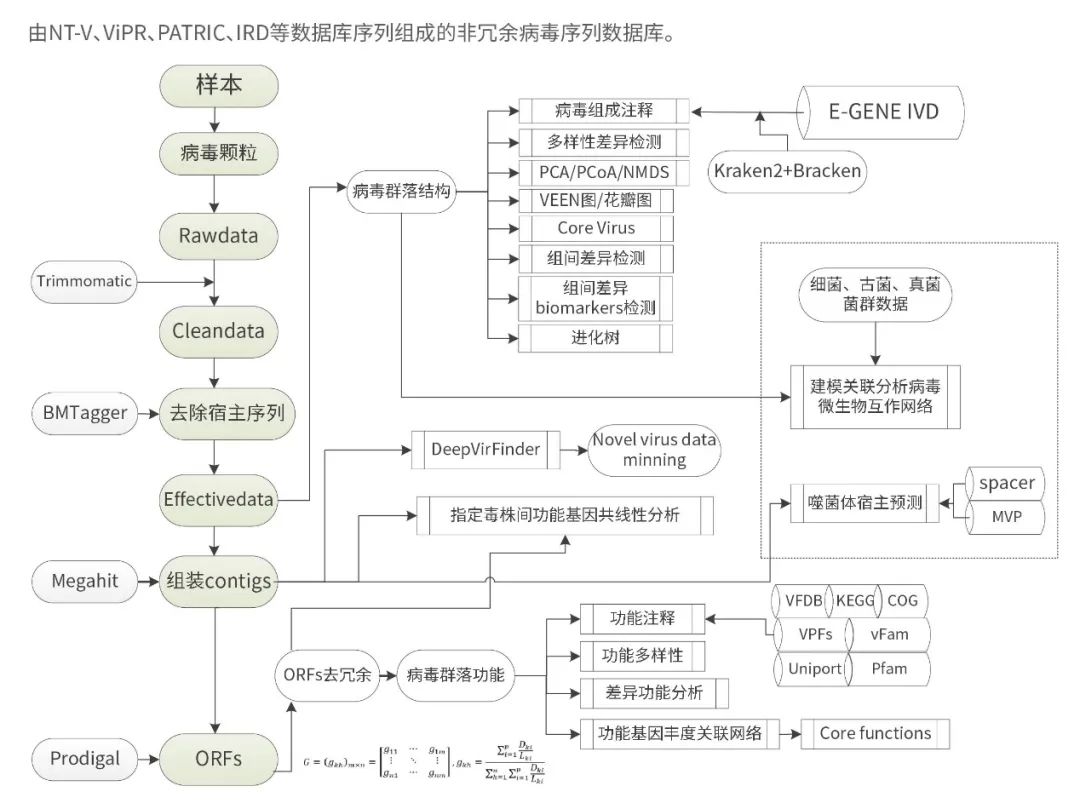
图1:EGENE 宏病毒组分析流程
测序数据质控和统计
宏病毒组的数据依然还是NGS测序平台下机的fastq格式的数据,因此在此部分大家可以放心使用常见的二代测序相关的质控及序列统计软件进行相应的工作。基础质控过滤,推荐使用Trimmomatic(version 0.38) (Bolger AM, et al. 2014),指控参数推荐为ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:8:true SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50。Clean后的数据及原始数据使用MultiQC(Ewels P, et al, 2016)对碱基测序质量进行评估,使用ReSeqTools对原始数据及clean数据进行数据产出统计。
去除宿主序列
虽然采用了病毒颗粒富集技术,但是并不能完全避免测得的数据中有宿主序列污染,获得Clean数据后,还需要对宿主序列信息进行剔除,去除宿主序列信息参考NCBI推荐的Human Sequence Removal SOAP,具体使用 程序为BMTagger。针对不同环境的样本构建宿主的相应数据库进行宿主序列去除。去除完毕后,对有效reads数据集进行相关指标统计,并统计宿主污染率。
样本病毒群落结构组成
病毒的分类与命名,经过长期的探索、研讨,迄今已有较大的发展,但现阶段的病毒分类,还不涉及精确的病毒的进化或系统发生。通用的病毒分类系统采用目、科、亚科、属和种的分类单元(分类等级)。病毒名称(英文)的分类学命名标准:
Kraken2是一个基于k-mer算法的高精度meta测序群落序列分类软件,能够快速的将测序reads进行物种分类。Bracken (Bayesian Reestimation of Abundance with KrakEN)( Lu J, et al, 2017)是一种基于贝叶斯算法的高精度统计方法,结合Kraken2可以实现高准确度的宏测序数据物种分类分析。目前病毒数据库有两大特点,一个是序列数目有限,并没有特别完善的综合性病毒数据库;一个是序列增长速度较快,要求要能够持续跟踪。易基因使用的病毒物种注释数据库为E-GENE IVD,该数据库为易基因科技综合NT数据库中的病毒序列、ViPR、PATRIC以及IRD的数据库中的病毒序列自主构建的非冗余病毒序列数据库。如果科研小伙伴们对病毒相关数据库比较感兴趣,后续小编可以就病毒数据库出一期专题前来介绍相关内容。
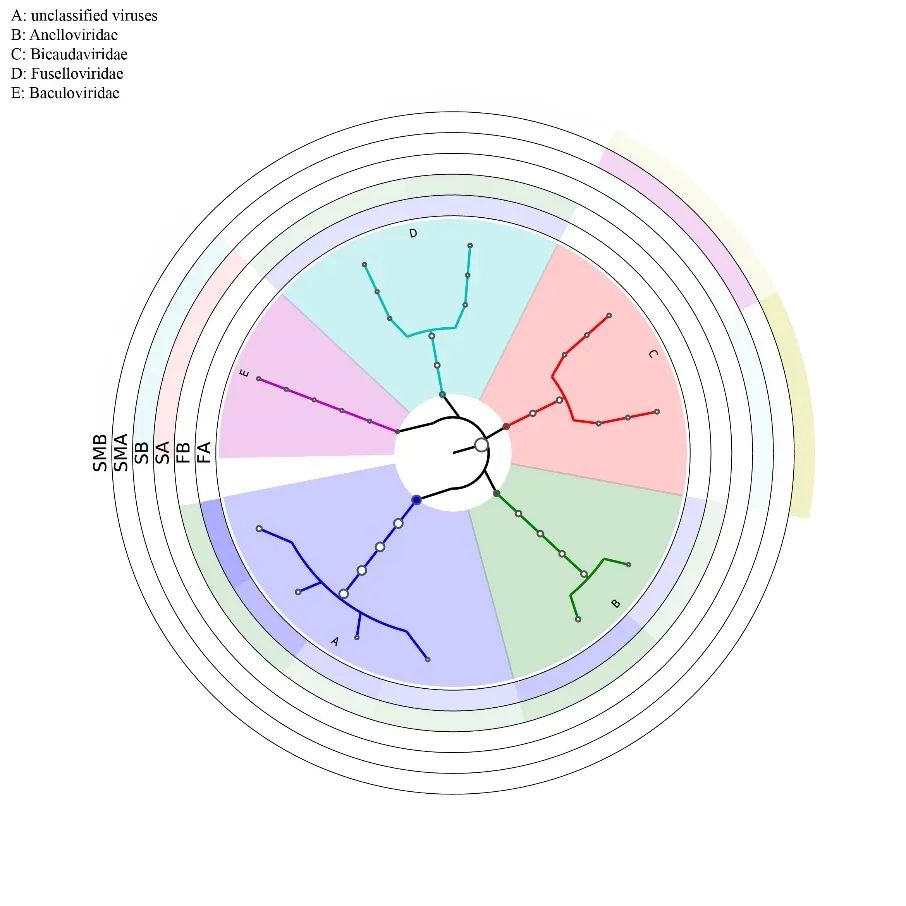
图2:病毒注释结果graphlan结构图
样本病毒群落结构多样性分析
Alpha多样性是指一个特定区域或生态系统内的多样性,是反映丰富度和均匀度的综合指标。Alpha多样性主要采用Shannon index(Che LQ, et al, 2019)和Simpson index对样本的Alpha多样性指数进行计算,并统计差异显著性,使用软件为R包vegan。不同于alpha多样性指数,Beta多样性用于不同生态系统之间多样性的比较,也就是样品间的差异。Beta多样性利用各样本序列间的进化关系及丰度信息来计算样本间距离,反映样本(组)间是否具有显著的微生物群落差异。主要使用Jaccard index和bray index来评估样本间的Beta多样性,利用的软件是R包vegan。利用Beta多样性指数进行非度量多维尺度(NMDS)分析。这些工作都可以通过R语言脚本轻松完成。
样本病毒群落结构组间差异分析
组学数据的组建差异分析软件较多,为了研究组间具有显著性差异的病毒种类,从不同taxonomy层级的病毒丰度表出发,利用 Metastats软件或者STAMP软件对组间的病毒丰度数据 进行假设检验得到 p 值,根据p值筛选具有显著性差异的物种,并绘制差异物种在组间的丰度分布箱图。如果是分组情况较为复杂,希望多组比较筛选组间具有显著差异的物种Biomarker,可以采用LEfSe软件(Hill NM, 2011)进行相关工作。首先通过秩和检验的方法检测不同分组间的差异物种并通过LDA(线性判别分析)实现降维并评估差异物种的影响大小,即得到LDA score;组间差异物种的LEfSe分析结果包括三部分,分别是LDA值分布 柱状图,进化分支图(系统发育分布)和组间具有统计学差异的Biomarker在不同组中丰度比较图。

图3:STAMP 差异病毒分析
单样本病毒序列组装
为了进一步对病毒基因层面深度解读,采用组装的方式获取更完整的序列。对于DNA病毒,综合考虑组装效果及计算资源消耗,推荐采用的软件为metahit,基础参数为–k-min 21 –k-max 149 –k-step 10 -m 0.3宏病毒怎么清除,组装完毕后对contigs进行过滤,去除200bp以下的contigs序列,每个样本独立组装,最终获得各个样本的有效组装序列。
病毒ORFs预测
我们采取的是基于ORFs注释的方法进行后续相关群落结构与功能的解析。这也是目前主流分析模式。
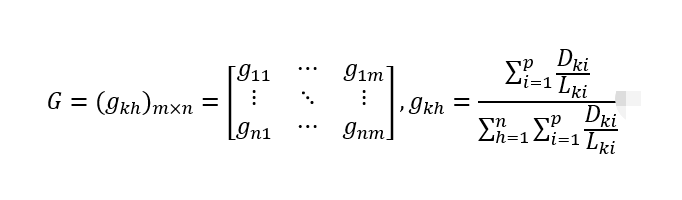
说明:m 为样本数, n 为代表性基因数目, gkh 为样本k的代表性基因h的标准化丰度,p为样本k的代表性基因h的总ORFs数目,Dki 为样本k包含的ORF i的reads数目,Lki为样本k的ORFi的序列长度。
耐药基因数据库CARD注释
功能注释这块针对最常见的KEGG及COG数据库的注释这块因为篇幅问题,不再做过多的介绍,如果科研小伙伴需要,评论区留言,小编可以满足大家做一期关于KEGG及COG数据库注释的专题哦。这里我们希望给各位小伙伴着重介绍一下抗生素耐药基因数据库的注释工作。随着抗生素药物的发现及使用,越来越多的耐药菌株由此产生。而耐药菌株的发展则会增加疾病治疗的难度和成本,因此耐药微生物的研究则显得尤为重要。虽然抗生素对病毒并无直接作用,但是有研究发现来自多种环境的病毒组携带着抗生素耐药性基因。这一结果提示着噬菌体—感染细菌的病毒—可能在转移让细菌产生耐药性的基因中发挥着作用。因此对病毒功能基因序列进行耐药性检测就显得尤为重要。目前,通过对耐药基因的鉴定挖掘能够一定程度上帮助我们揭开耐药机制,为疾病的治疗、药物研发提供参考。ARDB是最先整合了各种微生物中抗药基因的数据库,但它从2009年开始就不再更新。而CARD(the Comprehensive Antibiotic Research Database)数据库包含了ARDB数据库中所有抗性信息,并搭建了一个基于志愿者贡献的数据共享平台,做到了实时更新保证了数据的有效性。目前,CARD数据库收集了超过1600个已知的抗生素抗性基因。
CARD数据库()核心是ARO(Antibiotic Resistance Ontology), ARO包含了与抗生素抗性基因,抗性机制,抗生素和靶相关的term,如图所示。2017年发表的文章中,更新了数据库的相关功能,其中也提到了其他本体论,如用于描述抗生素抗性基因预测模块和参数的MO,定义不同term之间关系类型的RO,以及描述CARD中物种和菌株的NCBI Taxonomy。建议大家利用CARD官方软件rgi对全部得非冗余基因进行抗生素抗性基因注释。但注释之后如何批量的对注释的各个层面的注释结果进行提取、统计学分析以及可视化的工作,就靠大家各显神通通过自编脚本来完成啦。
噬菌体宿主预测
病毒基因组系统发育树
系统发育树(Phylogenetic tree,又称为系统发生树/系统发生树/系统演化树/进化树等),是用来表示物种间亲缘关系远近的树状结构图(Huelsenbeck JP, et al, 2001)。1965年,Linus Pauling等(Zuckerkandl E, et al, 1965)提出了分子进化理论,基于分子特性(DNA、RNA和蛋白质分子),推断物种之间的系统发生关系,由于核苷酸和氨基酸序列中含有生物进化历史的全部信息,因此利用该方法构建的系统进化树更为准确。在系统进化树中宏病毒怎么清除,物种按照亲缘关系远近被安放在树状结构的不同位置,因而,进化树可以简单地表示生物的进化过程和亲缘关系。病毒基因组系统发育分析为将目标基因组与近源序列一同建立进化树, 用以了解其系统发育关系, 将目标病毒与其他病毒的亲缘关系、 进化路径和聚类分群等情况进行可视化, 有助于梳理病毒之间的相关性和互作影响等关系。具体使用 blast 软件, 把目标基因组分别于E-GENE IVD数据库比对,使用 MEGA 软件构建进化树。
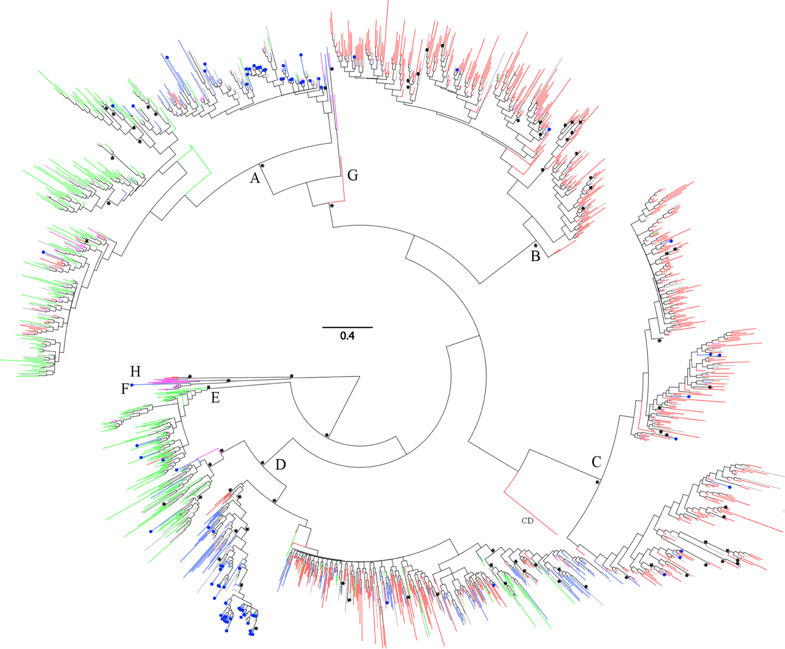
图4:病毒基因组系统发育树
病毒序列de novo预测
目前病毒序列的去除宿主以及注释识别的主要手段还是基因序列相似度比对的策略。但是目前微生物数据库并不完善,特别是病毒序列,数据库序列有限,通过比对注释识别效率不高。而DeepVirFinder(Ren J, et al, 2020)利用了深度学习和大数据的优势,无需与参考序列比对,显著提高了病毒识别的速度和准确性,将有助于在高通量组学时代下对病毒的研究。DeepVirFinder对基因序列搭建了基于卷积神经网络(convolutional neural networks)的模型,利用大量已知的病毒序列和细菌序列进行训练,得到了最优的二元分类器。卷积神经网络的优势在于它可以自主学习得到病毒的特征(motifs),无需事先定义,因此比传统的机器学习方法更加准确。另外,此模型利用已知序列学到了病毒的一般性特征,因此比基于序列比对的传统方法在识别未知病毒上更加灵活有效。
病毒-微生物互作关联分析
微生态环境中,病毒与细菌或者古菌之间的相互作用复杂。目前已知的先验知识并不充足,基于病毒丰度与细菌丰度表,利用统计学手段计算皮尔斯或者斯皮尔曼相关系数,构建病毒-微生物互作关联网络是有效深入解读病毒与其他微生物之间互作关系的有效手段。
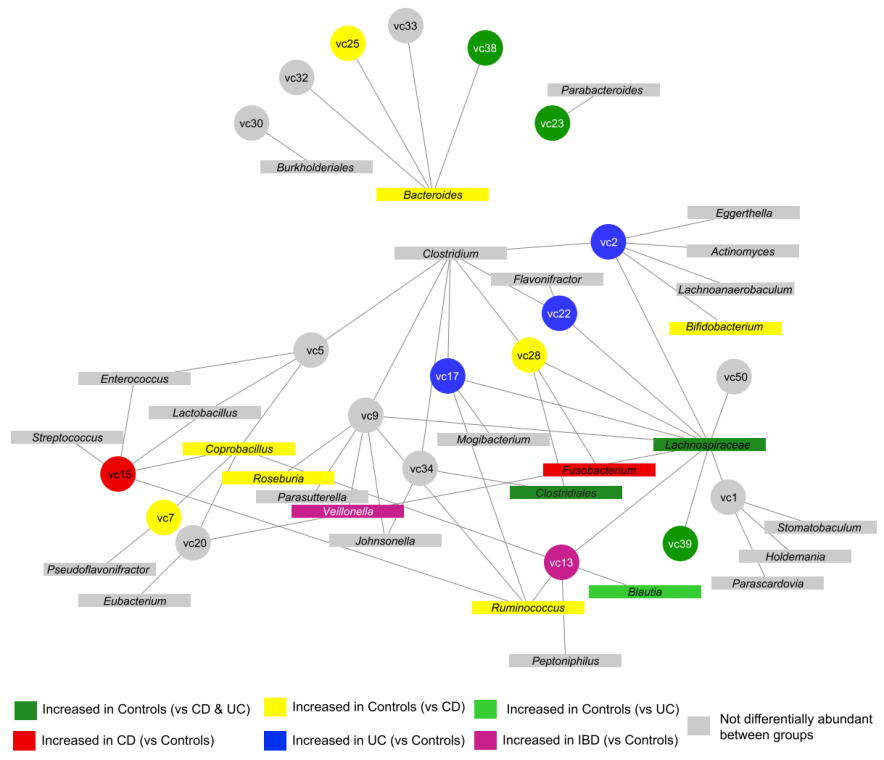
图4:病毒与细菌互作网络图
篇幅限制,
今天就先和大家聊到这里,
想跟踪了解最新的宏病毒组相关的分析进展及思路,
———END———
限 时 特 惠: 本站每日持续更新海量各大内部创业教程,一年会员只需98元,全站资源免费下载 点击查看详情
站 长 微 信: Lgxmw666